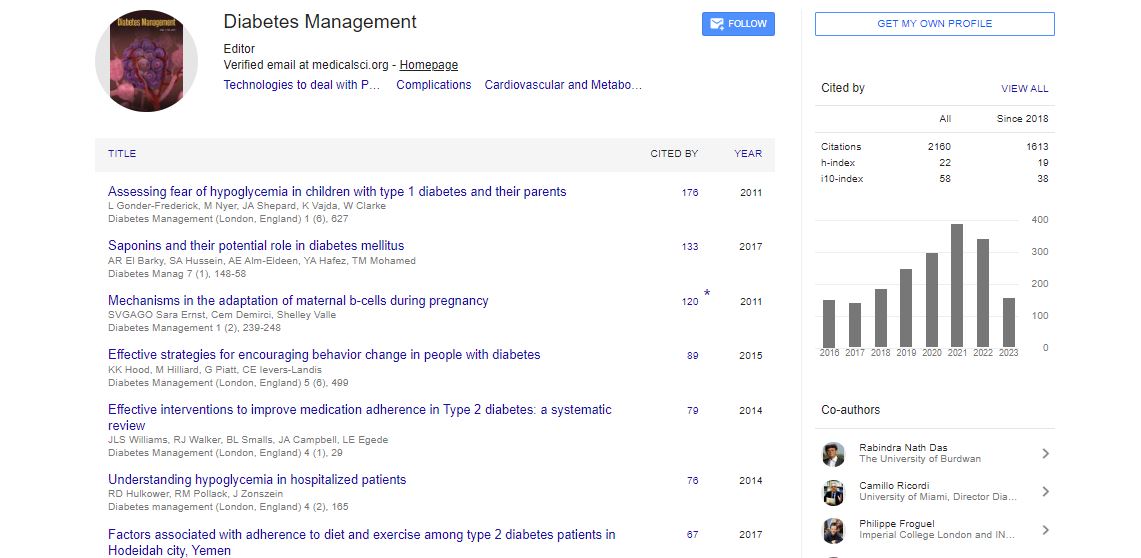
Research Article - Diabetes Management (2016) Volume 6, Issue 2
Cellular, Molecular and Therapeutic Advances in Type 2 Diabetes Mellitus
- *Corresponding Author:
- Pravin D. Potdar
Head of Department, Department of Molecular Medicine and Biology
Jaslok Hospital and Research Center
15 Dr. G. Deshmukh Marg, Mumbai – 400 026, Maharashtra, India
Tel: + 91-9820833530
E-mail: ppotdar@jaslokhospital.net
Abstract
Type 2 Diabetes Mellitus (T2 DM) is an ancient disease, discussed 3500 years ago in literature. Vast population worldwide is suffering from this disease and yet it is found to be untreatable. As per WHO statistics 2012, the number of deaths in the world due to T2 DM almost reached to 1.5 million. The number of people suffering from T2 DM in India is expected to rise from 40.9 million in 2007 to 69.9 million in 2025 due to change in their life style. Of the two types of DM- Type 1 and Type 2, the latter is most common, affecting 80% of DM patients. Modern lifestyle and food habits have been responsible for tremendous rise in the number of patients all over the world. Even though, the symptoms of T2 DM are seen in the later stages of life, the onset of the disease occurs quite early. Insulin resistance and β cell dysfunction are the main causative abnormalities. Several mutations in the genes important for glucose homeostasis and β cell development have been related to progress of hyperglycemia, while this progress is related to various metabolic syndromes arising in the patients’ body. Thus, forming a vicious circle of the causes and effects of hyperglycemia interlinked together forming the whole picture of T2 DM in the patient. This review discusses these causes and effects at the molecular and cellular levels. The current therapy practices use of oral therapeutic drugs, which controls hyperglycemia and related complications but fails to cure the disease permanently. Hence, stem cell therapy has drawn interest of the researchers in recent times, which seems to be a promising source of remedy for this notorious disease. This review will provide a better understanding of T2 DM and associated complications, recent advances in the therapy and the molecular and cellular insights of the disease to the students, clinicians, endocrinologists and diabetologists.
Keywords
Diabetes mellitus, stem cell therapy, molecular markers, metabolic syndrome, gene regulation
Introduction
Diabetes Mellitus (DM) is a disease known to humankind since a very long time. The disease has been mentioned in 3500 years old literature. The etymology of the name diabetes mellitus states its meaning as “the sweet flow or siphon.” In spite of being such an ancient disease a complete cure for DM is not available till date [1]. Millions of people all over the world are suffering from this disease; the incidence of DM is increasing year by year due to several reasons [1]. In India, 40.9 million people were suffering from DM in 2007. The number is expected to rise up to 69.9 million by 2025 [2]. The common symptoms of this disease are hyperglycemia, frequent urination, polyuria, polydipsia, and unexplained weight loss. Diabetic patients often show abnormalities termed altogether as “metabolic syndrome.” The indicative signs of this syndrome are high blood levels of triglycerides and low blood levels of high density lipoprotein (HDL) cholesterol which increase the risk of developing heart diseases in human [1,3]. Diabetes often leads to several complications such as stroke, myocardial infarction, loss of vision, renal failure and neuropathies. There are major two types of DM, Type 1 and Type 2. Type 1 Diabetes Mellitus (T1DM) is an autoimmune disease caused due to disturbance in T-cell mediated immune response of β cells of pancreas; whereas Type 2 Diabetes Mellitus (T2DM) is caused due to insulin resistance due to failure of β cell function. However, exact mechanism of insulin resistance is not yet known and further study in this area is needed. DM patients show impaired glucose tolerance, which is mainly caused by impaired insulin action and insulin secretory malfunction [4-6]. The predisposing factors are genetic susceptibility, environmental factors and lifestyle. The facets of modern lifestyle such as physical inactivity, sedentary habits, overly rich nutrition, cigarette smoking, consumption of alcohol and obesity have led to increase in spread of diabetes [7-9]. This review will overview major information about causation of T2DM at cellular and molecular level. It also highlights possible cure by advanced drug discovery program and stem cell therapy. This review will be very much useful for Post-graduate students working on DM as well as for clinicians to get a proper picture of T2DM so that they can plan precise treatment protocol for successful cure of this disease.
Mechanism involved in the onset and progress of T2DM
In humans, after glucose intake, normal glucose homeostasis mainly occurs through three major pathways. First, insulin is secreted by pancreas; second, decline in glucose production by liver cells; third, stimulation of glucose uptake by liver and peripheral muscles [10,11]. Shulman et al. [12] has further shown that muscle glycogen synthesis is the principal pathway of glucose disposal in both normal and diabetic subjects and thus defects in muscle glycogen synthesis plays very important role in the insulin resistance in T2DM. It has also been proved by Warrem et al. [13] that the symptoms of DM can be noticed one to two decades before the actual occurrence of this disease in offspring of diabetic patients. They have also shown that these patients have hyperglycemic state, which is recompensed by hyperinsulinemia. No hypoinsulinemia is observed at this stage thereby indicating that much before the loss of β cell activity the defect arises in peripheral tissue response to glucose. Hence, it can be stated that insulin resistance and not insulin deficiency leads to the onset of T2DM in these patients. T2DM patients show low concentrations of Glucose-6-phosphate and reduced rate of non-oxidative glucose metabolism. This indicates that either hexokinase or insulin stimulated glucose transport activity is reduced in patients. Rothman et al. [14] has shown that the reduction in insulin stimulated muscle glycogen synthesis may increase the risk of insulin resistance in patients with T2DM. This reduction in the rate of muscle glycogen synthesis indicates faulty glucose transport or hexokinase activity. They have further concluded that the offspring of parents with T2DM have defect in muscle glucose transport/hexokinase activity causing insulin resistance much before the onset of hyperglycemia and this may be primary factor in the pathogenesis of T2DM. Cline et al. [15] has also indicated that the defect in glucose transport/phosphorylation is the major factor responsible for reducing the rate of muscle glycogen synthesis in T2DM patients. The hyperglycemic state results in inflammation and stimulates β cells to produce Interleukin-1 (IL-1) at higher rate. The lower concentration of IL-1 antagonist fails to compete and bind the receptors. This results in β cell destruction and loss of function. Goyal et al. [16] has studied TNF-α and Interleukin-6 (IL-6) levels in obese and non-obese diabetic patients before and after insulin therapy and they found that there is increased inflammation in obese diabetic patients than in non-obese diabetic patients. They have further stated that to counteract this inflammation in obese patients, the response to insulin treatment is delayed than in non-obese diabetes patients.
Since completion of human genome project, several scientists are focused on finding out molecular mechanism of T2DM. Recently Saini et al. [17] has overviewed several such mechanisms involved in T2DM. Marino et al. [18] has studied major mechanisms involved in defective insulin stimulated glucose transport activity in DM patients. They have shown that there is an increase in intramyocellular lipid metabolites such as fatty acyl CoAs and diacylglycerol in DM patients. These molecules activate serine/threonine kinase cascade and lead to phosphorylation of insulin receptor substrate (IRS)-1 thus leading to defects in insulin signaling in these patients. They have further correlated reduction in mitochondrial function in the insulin resistant offspring of DM patients and thus by understanding these cellular and molecular mechanisms for insulin resistance there is a possibility of providing potential new targets for therapies of T2DM.
Akt plays a very important role in the insulinsignaling pathway via activation of insulin dependent glucose transporter protein GLUT4. Martin et al. [19] has shown that GLUT4 undergoes insulin dependent movement to the cell surface in adipocytes. These data suggest that, GLUT4 is selectively organized into a vesicular compartment in adipocytes and then transferred to the plasma membrane on insulin stimulation. Recent animal experimentations have shown that PKB (Akt2) isoform knockout mice exhibit insulin resistance and its phenotype closely resembles T2DM in humans. This explains that Akt2 is an essential factor in the maintenance of normal glucose homeostasis in humans [20,21]. George et al. [22] has suggested that inherited defects in signaling pathways may contribute to insulin resistance in human T2DM. The studies revealed that autosomal dominant inheritance of severe insulin resistance and DM in a family was related to mutations in kinase AKT2/PKB β domain. The insulin receptor substrate (IRS)- 1 is a major component of the insulin signal transduction pathway and mainly mediates both the metabolic and mitogenic responses to insulin [23]. Several investigators have examined the IRS-1 gene in insulin resistant T2DM patients and shown that more than 11 amino acids polymorphisms have been identified and mutations in the IRS-1 protein are associated with insulin resistance in human muscle and fat [24,25]. Kido et al. [26] has demonstrated in mice that IRS-1 and IRS-2 play important roles in the regulation and function of individual β cells and insulin resistance developed in these mice due to β cell failure. Studies have shown that serine phosphorylation of IRS proteins reduces the ability of IRS proteins to attract PI3- kinase resulting in reduction in activation and subsequent degradation of the IRS-1 protein [27-29].
Insulin resistance and pancreatic β cell dysfunction are major factors for cause of T2DM. It is also shown that there is reduction in β cell mass in severe DM. Nakae et al. [30] has shown that Foxo 1 gene is a negative regulator of insulin sensitivity in liver, adipocytes and pancreatic β cells. Recently Kitamura [31] has reviewed the role of Foxo1 in β cell dysfunction and DM. Potdar et al. [32] has shown that mesenchymal stem cell derived from peripheral blood of insulin resistant DM patient showed down regulation of Sox2 gene. This indicates that there is loss of pluripotency in these MSCs as well as they have further postulated that it may link to alteration of self-renewal properties of β cells.
Chronic hyperglycemia is a major cause of diabetic cardiovascular and microvascular complications, such as retinopathy, neuropathy and nephropathy [33]. It is further shown that increase in formation of diacylglycerol (DAG) and activation of protein kinase C (PKC) formed advanced glycated end products which are responsible for development of vascular complications in patients. Das Evcimen et al. [33] has shown that the activation of the DAGPKC pathway is associated with many vascular abnormalities in the retinal, renal, neural and cardiovascular tissues in T2DM. PKC isoforms, nPKCs plays major role in insulin signaling. Several studies have demonstrated a link between nPKCs and FFAs induced insulin resistance in DM patients [34,35]. Noh et al. [36] have reviewed the pathogenic role for the activation of DAG-PKC pathway in diabetic nephropathy and other microvascular complications in T2DM. Numerous mechanisms have been proposed to explain the association between hyperglycemia and vascular complications [37,38]. It is proposed to aim for an inhibitor of PKC isoform as a new therapeutics molecule, over viewing the involvement of PKC in diabetic vascular diseases, to delay or stop the progression of diabetic vascular complications in T2DM with minimal side effects.
Lifestyle, Obesity and T2DM
Many life style habits are responsible for prevalence of T2DM such as overweight, obesity, lack of physical activity, family history, Vitamin A deficiency, use of cholesterol lowering drugs, etc. [39]. We know diabetes and obesity interrelation from the earliest descriptions of the disease. The modern lifestyle and foodstuffs have resulted in an increase in obesity and thus diabetes cases. Most of the patients who are above 50 years age and overweight are suffering from T2DM [7]. Obesity is an important factor linked to DM however, exact relation and mechanism is inadequately understood. Ozcan et al. [40] has worked to show that obesity leads to endoplasmic reticulum stress, which plays an important role in peripheral insulin resistance and hence in occurrence of T2DM at the molecular, cellular, and organismal levels. Fiber and fat content of the diet, alcohol consumption, smoking habit and sedentary lifestyle play a significant role in prevalence of DM. A higher incidence of DM is linked to Body Mass Index higher (BMI) than 25 kg/m2. The results are similar worldwide as reported in studies from all over. The association between BMI and DM occurrence is found to be stronger among younger, than among older individuals [41-43]. A research on Pima Indians reveals that incidence of DM and obesity has increased in this century. This is possibly due to the changes in lifestyle and diet of the people who are already genetically susceptible to diabetes. The risk is higher when one of the parents is diabetic and even higher when both the parents are diabetic [8].
The current therapies like insulin, diet, exercise, behavior modification, and oral agents, fail to return patients to long-term euglycemia. Gastric bypass seems to be a promising option to restore and maintain normal levels of glucose, insulin, and HbA1c. It also reduces considerable amount of body fat. The possible reasons for the success of gastric bypass could be- total caloric intake is limited, proportion of carbohydrates in the diet is reduced, food is excluded from the hormonally active parts of the digestive system i.e. antrum, duodenum, and proximal jejunum, transit time from the stomach to the small intestine is delayed due to the small gastric outlet. This operative procedure can also prove to reverse other related complications like cardiopulmonary function, cure sleep apnea and snoring, control asthma, clear peptic reflux, improve physical activity of patients suffering from arthritis, and restore fertility [44]. Lifestyle changes along with proper medications can prove to control morbidity and mortality of diabetes and related metabolic syndromes.
Recent study by Imamura et al. [39] has shown that regular consumption of sugar-sweetened drinks, fruit juice or artificially sweetened drinks may have the risk of T2DM. They have further suggested that sugary drinks are associated with greater risk of obesity, which is a major risk factor for T2DM. In general, fruit juice and artificially sweetened beverages are recommended for good health. However, it is not clear whether, consuming such drinks can be blameless for occurrence of T2DM because these factors are responsible for development of obesity and eventually responsible for developing DM. Li et al. [45] have found that birth weight along with lifestyle in adulthood is related to T2DM. They have suggested that prenatal and postnatal factors are both responsible for causing DM. In addition, it has been shown that unhealthy lifestyles such as smoking, irregular and unhealthy diet, less physical activity, alcohol consumption and high BMI are responsible for causation of obesity and DM in human. The study concluded that T2DM could be prevented by adoption of healthier lifestyle and improvement in prenatal and postnatal factors.
Role of NF-κB and NF-κB Regulated Cytokines in T2DM
NF-κB is a transcription factor, which plays an important role in many biological processes. It regulates the expression of various cytokines, growth factors and other molecules. There are five different types of NF-κB proteins- p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1) and p52/ p100 (NF-κB2). These proteins are inhibited or arrested in inactive state by Inhibitor of NF-κB (IκB) in cytoplasm in normal condition. IκB prevents translocation of NF-κB to nucleus. Protein IκB kinase (IKK) phosphorylates IκB, thereby leading to the degradation process of IκB protein and hence NF-κB is activated. This occurs in several disease conditions [46]. Recent studies have shown that NF-κB and NF- κB regulated cytokines play significant role in development of T2DM [47,48].
NF-κB levels are elevated in obese and diabetic patients. The accumulation of FFAs in liver causes inflammation by NF-κB pathway and release of cytokines, which eventually leads to insulin resistance [49,50]. Arkan et al. [51] has demonstrated the importance of IKK in insulin resistance and also the role of NF-κB as the main causal factor in prevalence of DM. The higher concentration of IKK leads to over activation of NF-κB. This could be reversed by salicylate inhibition of IKK [50]. Obesity brings about alteration in expressions of genes of hormones and cytokines in adipocytes. TNFα is an adiponectin which plays a crucial role in lipid metabolism and insulin resistance [52,53]. TNFα is also associated with BMI, body fat distribution and β cell dysfunction due to autoimmunity and cytokine production [54,55]. This results in elevation of FFAs concentration by stimulation of lipolysis, and negative regulation of important insulin sensitizing nuclear receptor, Peroxisome proliferator-activated receptor gamma (PPARG). Thus, neutralization of TNFα could possibly be helpful in reversing the effects of insulin resistance and preventing β cell apoptosis [56,57]. High plasma levels of C-reactive protein (CRP) and IL-6 also contribute to inflammation and insulin resistance. Insulin resistance caused due to IL-6 was shown to be reversed by systemic neutralization of IL-6 in mice [50,53,58]. IL-1 receptor antagonist is reduced in T2DM and therefore increases in IL-1 concentration, resulted due to high glucose level, brings about β cell dysfunction and hence affects insulin regulation. Larsen et al. [59] has stated that blocking IL-1 with anakinara helps to control hyperglycemia, improve β cell functions and insulin sensitivity and overcome inflammation. Liu et al. [60] has mentioned about the upregulation of IL-1 in patients with diabetic retinopathy. The role of NF-κB and NF-κB regulated cytokines have also been mentioned in prevalence of diabetic nephropathy [61,62], diabetic cardiomyopathy [48,63] and diabetic retinopathy [60,64]. Chronic activation of inflammatory pathways in skeletal muscle is one of the major factors in the pathology of obesity, insulin resistance and T2DM [65]. More than 75–80% of glucose disposal in humans is through skeletal muscle and therefore impairment of insulin action in this tissue is major site for insulin resistance. Of late, Green et al. [66] has shown that NF-κB signaling serves as an indicator of inflammatory signaling pathway in skeletal muscle. Overall, it suggests that control of NF-κB and NF-κB regulated cytokines may prove to be a useful treatment for T2DM.
Role of Mitochondrial DNA in T2DM
Insulin is stored in secretory vesicles in β cells and is released on glucose stimulation by exocytosis as shown in Figure 1. Mitochondrial functions are essential for this glucose stimulated insulin secretion. Mitochondrial dysfunction and mutations in mitochondrial DNA (mtDNA) leads to impaired insulin secretion and T2DM [67,68]. mtDNA consists of two regions, coding region which codes for RNAs and control region which controls the expression of the mtDNA [69]. Variants arise due to mutations in any of these regions in the DNA during evolution [70]. Also, the weak repair mechanism in mitochondria is inefficient as compared to the nuclear DNA repair mechanism and hence allows transmission of these mutations at a higher rate [69,71].

Figure 1: Role of mitochondria in insulin activation.
mtDNA is inherited only from the mother and thus the mutation occurred in mtDNA can only be transmitted via mother to child. Approximately 1% of all diabetes patients are suffering from maternally inherited diabetes and deafness (MIDD) which is a rare form of diabetes caused due to specific mtDNA mutation. This mutation was first discovered by Van den et al. [72] in 1992 in a Dutch family and by Reardon et al. [73] in a UK family. MIDD is mainly caused due to A to G substitution at the conserved position 3243 (m.3243A>G) of the mtDNA and so far, it is investigated by several investigators [74-76]. Maternal mitochondria are inherited by the offspring and thus the prevalence of T2DM could be attributed to maternal inheritance [77]. Recently Crispim et al. [78] has investigated the prevalence of ten described mtDNA mutations in MIDD patients from Brazil. They have also studied most common A3243G mutation in the tRNA Leu (UUR) gene in these patients along with other 9 mutations such as C1310T, A1438G, T3271C, G3316A, T3394C, A8344G, A12026G, T14577C and T14709C. Fania et al. [79] has studied four regions of mtDNA- the tRNA Leu (UUR) gene, NADHdehydrogenase subunit 1 (ND1) gene and part of the 16S-rRNA gene and ATPase8 gene. Thus, they have stated the importance of mtDNA mutations in the pathogenesis of MIDD and other T2DM patients.
A common mitochondrial variant called 16189 variant is a result of T to C substitution at nucleotide position 16189 in the hypervariable D-loop (control) region of mtDNA [80,70]. Ye et al. [81] has reported in their review on Europid population study that Ori B variant i.e. 16181-16193 polycytosine variant, of mtDNA augments the probability of prevalence of T2DM. Tawata et al. [76] has mentioned about another mutation in mtDNA at 14577 T/C which may probably be an important cause of maternally inherited T2DM. Liu et al. [82] has found novel mtDNA mutations in MIDD in Chinese population. Similarly Tang et al. [83] has shown variation in mtDNA mutations associated with T2DM in a Chinese population.
Due to these inherited and acquired defects, obesity and T2DM patients show impaired mitochondrial function in their skeletal muscles [84,85]. This results in accumulation of FFAs, which causes reduction in insulin stimulated muscle glucose transport. This leads to fall in muscle glycogen synthesis and glycolysis [35,86]. Defective insulin stimulated glucose transport is an early event in the pathogenesis of T2DM [87].
Molecular Markers in T2DM
Occurrence of T2DM is associated with alteration in expression of a number of genes. Detection of these alterations in the gene expression is useful in revealing the stage of disease in the patients and provides a better understanding for its treatment.
▪ Endothelial dysfunction related molecular markers in T2DM
One of the major complications caused due to T2DM is the development of cardiovascular disease in human. The endothelium plays a very important role in the regulation of production of vasodilator molecules such as Nitric Oxide (NO), prostacyclin, endothelium derived hyperpolarizing factor (EDHF), and vasoconstrictors such as Endothelin-1 (ET- 1) and angiotensin II for maintenance of arterial quality and proper blood flow [88]. Endothelial dysfunction is related to insulin resistance, inflammation, atherosclerosis and cardiovascular disease in T2DM [89-91]. Thus, the expression of molecular markers released by endothelial cells can provide us an estimate about endothelial dysfunction in these patients. Loss of bioavailability of NO, overproduction of vasoconstrictors and imbalanced regulation of inflammation, thrombosis, and cell growth in the vascular wall are the major signs of endothelial dysfunction [88,92]. Numerous studies have confirmed that the dysfunction of endothelium causes development of atherosclerosis and insulin resistance in obese and T2DM patients [93-95].
In normal condition, proper regulation of NO by endothelium prevents leukocyte adhesion and inflammation of arterial wall [88]. However, dysfunctional endothelium enhances the pathogenesis of atherosclerotic vascular disease. This results into inflammation and thrombosis of arterial wall which interfere in regulation of arterial tendency and flow [91,96]. In these conditions, the dysfunctional endothelium is activated to express vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), soluble E-selectin (sEselectin), chemotactic factors, such as monocyte chemoattractant protein-1 and cytokines such as macrophage colony-stimulating factor, and tumor necrosis factor-beta (TNF-β), which help in the adhesion of leukocytes to the endothelial surface [97-99]. An increase in prothrombotic factors like Plasminogen activator inhibitor-1 (PAI-1) by endothelial cells is observed in T2DM [90]. PAI-1 causes accumulation of extracellular matrix in kidneys thereby leading to diabetic nephropathy [100]. Upregulation of PAI-1 and fibrinogen causes thrombosis and inefficient clots dissolution. Insulin antagonizes platelet activity by sensitizing the platelet to prostacyclin and NO secreted by vascular endothelium [101]. In T2DM patients, this activity becomes faulty due to lack of insulin. As a result, platelets adhere to vascular endothelium and aggregate more readily, contributing to faulty macrovascular and microvascular events [102]. Overall, it is clear that endothelial dysfunction is very well linked to cardiovascular problems in T2DM patients.
▪ Cytokines and chemokines markers in T2DM
Cytokines are low molecular weight polypeptides having autocrine, paracrine, and juxtacrine activity. They are classified into several different types such as interleukins, tumor necrosis factors, interferons, colony-stimulating factors, transforming growth factors and chemokines. Cytokines are produced by a wide variety of cells in the body and also have great therapeutic potential [103]. Cytokines are involved in pathogenesis of DM and its microvascular complications. Inflammatory cytokines like IL-1, IL-6, IL-18 and TNF-α play major role in development and progression of diabetic nephropathy [107]. Inflammatory proteins are other important molecular markers of T2DM. Elevated levels of IL-6, TNF-α, TGF-β and CRP are stated to be involved in development of T2DM [58,104,105]. High production of IL- 1β is induced in β cells due to hyperglycemia, resulting in faulty insulin secretion, deficient cell proliferation, and apoptosis [59]. Moriwaki et al. [106] has discussed elevated levels of IL- 18 along with that of IL-6 and TNF-α in the serums of T2DM patients. On the other hand, it is observed that the serum concentration of anti-inflammatory proteins like adiponectin and IL-10 is reduced in obese and diabetic patients [107,108].
Irisin and other molecules as markers for obesity and T2DM
A recent discovery has introduced Irisin as a molecular marker in obesity, metabolic syndrome and T2DM. Irisin level is lower in T2DM patient serum compared to that in non-diabetic person [109]. Irisin is an exercise-mediated myokine, which controls energy metabolism by inducing browning of white adipose tissue and thus converts chemical energy to heat energy. Bostrom et al. [110] has reported amelioration of glucose levels and obesity induced by Irisin in humans and mice. Exercise induced mild increase in Irisin levels was shown to boost energy expenditure in mice. Thus, Irisin controls diet induced weight gain and its effects like obesity, insulin resistance and T2DM. The studies on caveolin-1 (CAV-1) gene expression in obese and diabetic patients by Catalan et al. [111] show that there is an upregulation of CAV-1 in the patients. CAV-1 is known to play important roles in many facets of cellular biology, such as vesicular transport, cholesterol homeostasis and signal transduction. This study implies that CAV-1 probably has some function in prevalence of obesity and obesity associated T2DM. Lancha et al. [112] has reported upregulation of syntaxin 8 (STX8) in Visceral adipose tissue of obese T2DM patients. STX8 is an important protein in endosome transport mechanism. The study suggests that high expression of STX8 may possibly be associated with glucose transport mechanism and occurrence of T2DM in obese patients. Visfatin or pre-B cell colony enhancing factor (PBEF) and apelin are adipocyte secreted hormones highly expressed in visceral fat and blood of obese patients. Visfatin promotes the growth of B cell precursors and acts as an insulin analog on the insulin receptor. Studies have reported that visfatin and apelin levels could be associated with occurrence of T2DM [113,114]. Some studies have investigated the role of mutation in Calpain-10 gene in development of T2DM. Calpain- 10 is a ubiquitously expressed member of the calpain-like cysteine protease family. Calpain-10 is thought to be involved in glucose metabolism [115,116]. Further study of the function of these genes and their products is required to obtain a better understanding of their roles in glucose homeostasis. Mutations in certain other genes crucial for development and functioning of pancreas and liver have been shown to be responsible for development of T2DM. NEUROD1, PDX-1, Hepatocyte nuclear factor-1 homeobox A (HNF1A), Hepatocyte nuclear factor- 1 homeobox B (HNF1B), Insulin receptor substrate-1 (IRS- 1), Insulin receptor (INSR) and High Mobility Group A1 (HMGA1) are such essential genes associated with progression of T2DM [117,118]. However, further investigation is necessary to conclude the susceptibility of these genes in development of T2DM in human.
Complications Associated with T2DM
The complications associated with T2DM intensify the state of morbidity and mortality of the patients. Diabetes complications are of two types- microvascular, which are caused due to damage to small blood vessels and macrovascular, which are caused due to damage to larger blood vessels. Microvascular complications include retinopathy leading to blindness, nephropathy leading to renal failure and neuropathy leading to impotence and diabetic foot disorders including severe infections, which may even lead to amputation. Macrovascular complications include cardiovascular diseases like heart attacks, strokes and deficiency of blood flow to legs [119]. We have further discussed some of these complications and their association with incidence of T2DM.
▪ Retinopathy
Diabetic retinopathy is a very common microvascular complication associated with diabetes, leading to blindness in a large number of people suffering from diabetes per year [120]. It is a chronic ocular disorder developing, to some extent, in almost all diabetic patients as the disease progresses. The common characteristics of diabetic retinopathy are retinal non perfusion, wherein the blood supply to the retina ceases because of alterations in microvasculature causing lesions in retina. In addition, increase in vasopermeability, capillary degeneration, uncontrolled neovascularization and macular edema is also observed [121]. Maintenance of euglycemia and control on blood pressure helps in delaying diabetic retinopathy and slowing its progress. Procedures used for treating diabetic retinopathy are cryotherapy, virectomy and laser photocoagulation. The recent treatment procedures include injection of steroid triamcinolone and vascular endothelial growth factor (VEGF) antagonists into the eye [121,122].
▪ Nephropathy
Nephropathy develops in 30-40% of diabetes patients and the main causal factor is hyperglycemia. Other factors responsible are hyperlipidemia, hypertension, and proteinuria. Diabetic nephropathy is characterized by persistent microalbuminuria, hyperfiltration and renal lesions in patients. In the earlier stages of the disease, accumulation of extracellular matrix in the mesangium and glomerular basement membrane is observed. As the disease progresses, mesangial nodules are formed and tubulointerstitial lesions are seen [123,124]. The symptoms of the disease in earlier stages can be reversed by insulin treatment [123]. The best treatment approach for diabetic nephropathy is pancreas transplantation [124,125]. Angiotensin-II-receptor antagonist such as losartan and angiotensin converting enzyme (ACE) inhibitors are used for therapy. However, patients suffer from the side effects of these drugs [126,127].
▪ Neuropathy
The peripheral nerve dysfunction observed in diabetic patients is called diabetic neuropathy [128]. It affects different parts of the nervous system. The most common diabetic neuropathies are Diabetic Peripheral Neuropathy (DPN) and Diabetic Autonomic Neuropathy (DAN). DPN patients suffer from loss of reflexes and sensations, especially in their lower limbs. They experience burning, pricking, electrical, stabbing or tingling sensations. Some patients experience extreme numbness. Almost half of the DPN cases are asymptomatic. These patients are at risk of developing ulcers, which may even lead to amputations. DAN may affect any of these organs- heart, lungs, blood vessels, bone, adipose tissue, sweat glands, gastrointestinal system and genitourinary system. The patients suffer from gastroparesis, constipation, diarrhea, anhidrosis, bladder dysfunction, erectile dysfunction, resting tachycardia, silent ischemia, and sudden cardiac death [129,130].
Like earlier discussed complications, diabetic neuropathies also develop as diabetes progresses. The accurate pathophysiology of diabetic neuropathies is unknown but is probably related to polyol accumulation, generation of Advanced Glycation End (AGEs) products and Reactive Oxygen Species (ROS) [131].
There is no cure to reverse diabetic neuropathy till date but the symptoms of the disease can be treated [129,131]. Therapeutic drugs are prescribed to the patient depending upon the symptoms shown by the patient and the type of neuropathy implicated. Anticonvulsants or antiepileptic drugs like gabapentin, pregabalin, topiramate carbamazepine, and lamotrigine are employed to treat painful neuropathies [129,132,133]. Other anticonvulsants being analyzed for their effectiveness are clonazepam, valproate, tiagabine, oxcarbazepine and levetiracetam [134]. Antidepressant drugs like desipramine, imipramine, nortriptyline and amitriptyline block norepinephrine uptake and hence have analgesic effect [133,135]. Acupuncture and electrical stimulation are other effective treatment procedures [133]. There is no single efficient treatment for diabetic neuropathy; various procedures discussed are utilized in synergy to give the patient a successful treatment.
Although the treatment procedures used to treat these disorders are effective, they have certain side effects. In addition, these procedures do not cure the diseases but only heal the symptoms. The best cure for these complications associated with DM is lowering the blood sugar level and blood pressure. Maintenance of euglycemia and normal blood pressure helps in delaying these complications and slowing their progress.
Therapies of T2DM
▪ Oral therapeutic agents
The main goal of therapy of DM is to maintain euglycemia. Maintenance of normal blood glucose levels reduces risks of related metabolic syndromes. Physicians prescribe a particular drug keeping in mind several aspects and their adverse effects. Various oral antihyperglycemic agents are equally efficient in lowering blood glucose concentrations and HbA1c, except for the Alpha glucosidase inhibitors (AGIs) and nateglinide. Sulfonylureas result in reduction of microvascular complications, while metformin result in reduction of microvascular as well as macrovascular risks. Insulin sensitizers, metformin and TZDs, avoid the possibility of hypoglycemia associated with secretagogue therapy [4]. Majority of patients suffering from T2DM require multiple therapy to attain normal glycemic levels, as monotherapy rarely serves the purpose and if it does, it takes comparatively very long time [4,136]. These therapeutic agents provide a control over hyperglycemia; however they are not efficient in curing the disease and related complications permanently. In addition, they cause several side effects. Recent developments in stem cell therapy would provide a better solution over these problems. In fact, the disease can be permanently sorted out by this approach. Also, unlike organ transplantations, stem cell therapy does not face problems like graft rejection and shortage of donors. The patients’ own stem cells can be differentiated in vitro to islet cells and these autologous cells can serve the purpose for curing this disease [1,6].
▪ Stem cells therapy a new avenue for treatment of T2DM
Stem cells have enormous potential to divide and differentiate into various different types of specialized cells of the body. Thus, stem cells therapy is a promising key not only for treatment of T2DM but also for many other disorders and diseases for which complete cure is not yet available. Stem cells could prove to be a non-exhausting source of the degenerated β cells in T2DM patients [6]. Figure 2 illustrates major causes of T2DM in human. There are two major faults associated, one is impairment of insulin function and other is β cell dysfunction [137,138].

Figure 2: Major causes of T2DM in humans.
Both the aspects of β-cell physiology can be comprehensively corrected with stem cell therapy. β cells can be cultivated from stem cells and used further for transplantations in diabetic patients. Research is going on for developing and establishing functional β cell mass from different types of stem cells. Recent progress in research raises hopes for T2DM patients to be cured in future by this miraculous therapy.
Human embryonic stem cells: Human Embryonic Stem Cells (HESCs) are the cells derived from the early human embryo and can propagate indefinitely in the undifferentiated state while exhibiting pluripotency. They are capable of giving rise to cells derived from all three germ layers. Thus, these cells can be cultured in vitro and further differentiated to provide a source of various specialized cells that can be used for replacement therapies of degenerative diseases [139]. Barclay et al. [140] has described a study where ESCs were successfully used in concert with oral therapeutic drugs like metformin or sitagliptin to treat T2DM in mice. Kroon et al. [141] derived pancreatic endoderm from hESCs which could differentiate into glucose-responsive, insulinsecreting cells in vivo. D’Amour et al. [142] has derived insulin, glucagon, somatostatin, pancreatic polypeptide and ghrelin secreting pancreatic endocrine cells from hESCs. The cells were differentiated by process designed to imitate the in vivo development process of the islet cells. The study of Cai et al. [143] has revealed that NGN3 expressing cells characterize pancreatic endocrine progenitor cells. These cells express PDX1, NKX6.1, and chromogranin A and also differentiate in vivo to insulin, glucagon, and somatostatin producing cells. The cells thus obtained from hESCs express β cell markers and exhibit their functions too [141,144]. ESCs are less immunologically potent than allogenic adult stem cells and hence may require less immune suppression [6]. However, ESCs stand the risk of developing into teratomas from the undifferentiated fraction of the transplanted cells. The other limitation of the use of ESCs is the ethical issues involved and therefore use of hESCs is restricted in stem cell therapy.
Mesenchymal stem cells: Mesenchymal Stem Cells (MSCs) can be obtained from various body tissues, viz. bone marrow, adipose tissue ,umbilical cord or its blood , fibroblasts , endometrium, and liver cells; bone marrow being the richest source [138,145,146]. Bone marrow derived MSCs are highly capable of self-replicating and differentiating, both in vitro and in vivo, to various tissue cells. They are easy to cultivate and remain pluripotent for long durations of culturing. They maintain the capacity of multilineage differentiation [6]. Jiang et al. [145] has reported that the multipotent adult progenitor mesenchymal cells proliferate extensively without any apparent senescence or loss of differentiation potential, and hence they may be an ideal cell source for therapy of inherited or degenerative diseases such as diabetes. Bone marrow derived MSCs transplantation in concert with hyperbaric oxygen treatment is found to be effective in improvising metabolic variables such as fasting plasma glucose, C-peptide, HbA1c and C-peptide/glucose ratio, as well as reduction in insulin requirements [147].
Bone marrow MSCs can be effectively used to produce glucose induced insulin-secreting cells, arrest insulin resistance, recovery of liver and pancreas damage and to cure diabetic ulcers and limb ischemia. MSCs transplantation also results in elevation of GLUT4, insulin receptor substrate 1 (IRS-1) and Akt (protein kinase B) [148-150]. MSCs were shown to suppress diabetogenic T cells and thus prevented autoimmune β cell destruction and other related disorders in mice [151]. Zhao et al. [152] worked on Bone marrow MSCs to study their effect on pathogenic action of hyperglycaemia in T2DM. Their findings reveal that there was an improvement of β cell function and survival induced by MSCs mediated through autophagy; also the alterations caused due to hyperglycemia were reversed. An alternative source of stem cells, peripheral blood could provide a more non-invasive approach. The mesenchymal stem cells thus obtained from the peripheral blood can be used in future regenerative stem cell therapy [32]. The effects of single MSCs transplantation are relatively short. In order to obtain long term maintenance of euglycemia, multiple intravenous transplantations must be carried out. Multiple infusions effectively maintain normal glucose levels and ameliorate the pathogenic effects of hyperglycaemia [153,154]. MSCs are the most studied type of stem cells for DM therapy. However, the safety and efficacy of MSCs therapy needs to be confirmed yet so as to use this approach for treatment.
Induced pluripotent stem cells: Induced Pluripotent Stem Cells (IPSCs) are developed from differentiated adult somatic cells by establishing expression of early development genes Oct3/4 and Sox2, along with cMyc and Klf4 or Nanog and Lin28. These embryonic-like stem cells show high clonogenicity and are able to give rise to cell lineages from all three germ layers. This technology can hence be exploited to produce fully functional β cells [155-157].
Jeon et al. [158] illustrated that IPSCs cultured from non-obese diabetic mice show typical ESCs-like characteristics such as expression of markers for pluripotency, in vitro differentiation, teratoma formation, and generation of chimeric mice. It was also demonstrated that epithelial cells derived from the pancreas of the nonobese diabetic mice differentiated more readily into insulin-producing cells and expressed various pancreatic β cell markers. These cells also secreted insulin in response to glucose and KCl stimulation. Transplantation of these cells into diabetic mice led to kidney engraftment. The engrafted cells successfully normalized blood glucose levels by insulin secretion. Thus, patient-derived IPSCs can prove to be a potential therapy for autologous β cell transplantation for treating diabetes. Alipio et al. [159] successfully achieved long term amelioration of hyperglycemia in mice by transplanting iPSC derived pancreatic β -like cells. The cells obtained efficiently secreted insulin in response to glucose stimulation and lowered blood glucose and hemoglobin A1C levels.
T2DM is a cosmopolitan disease and yet incurable in spite of long periods of research and efforts. Tissue regeneration and stem cell therapy is a ray of hope for the treatment for this wide spreading disease. However, the safety issues involved must be considered and further advancements in the protocols are required before clinical application of this study so as to provide patients an effective long term treatment. The efficacy and mechanism of stem cells also needs to be determined so as to understand study well and make new researches to develop better treatment.
Conclusion
T2DM being an ancient disease, cure of this disease has not been possible due to lack of study of the various mechanisms causing this disease. Due to advancements in Molecular Medicine and Stem Cell Research, there are hopes for the use of these advance technologies for complete cure of T2DM. This review has mainly highlighted the important mechanisms involved in occurrence of T2DM, Molecular profiling, Insulin resistance, endothelial dysfunction and related Molecular Markers. There is a probability of developing stem cell therapies by using MSCs or IPSCs cells for autologous transplantation for complete cure of this disease. This review will be of importance for future studies to be carried out in this area by researchers, clinicians, endocrinologists and diabetologists to define T2DM precisely and discover some key for the treatment of T2DM in humans.
Acknowledgement
Authors are thankful to Management of Jaslok Hospital and Research Centre for providing grant to do research on stem cells and related diseases. The experience gained during this program allows us to write this review article. We are also thankful to Mrs. Naina Rane for helping us in editing of this manuscript.
References
- Patlak M. New weapons to combat an ancient disease: treating diabetes. J. Faseb. 16(14), 1853 (2002).
- Mohan V, Sandeep S, Deepa R, Shah B, Varghese C. Epidemiology of type 2 diabetes: Indian scenario. Indian. J. Med. Res.125(3), 217-230 (2007).
- Olokoba AB, Obateru OA, Olokoba LB. Type 2 diabetes mellitus: a review of current trends. Oman. Med. J. 27(4), 269-273 (2012).
- Inzucchi SE. Oral antihyperglycemic therapy for type 2 diabetes: scientific review. JAMA. 287(3), 360-372 (2002).
- Lillioja S, Mott DM, Spraul M et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N. Engl. J. Med. 329(27), 1988-1992 (1993).
- El-Badri N, Ghoneim MA. Mesenchymal stem cell therapy in diabetes mellitus: progress and challenges. J. Nucleic. Acids. 2013, 194858 (2013).
- Marble A, Krall LP, Bradley RF et al. Joslyn's Diabetes Mellitus. Philadelphia: Lea and Febiger 1985, 373-379, (2005).
- Knowler WC, Pettitt DJ, Savage PJ, Bennett PH. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am. J. Epidemiol. 113(2), 144-156 (1981).
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 414(6865), 782-787 (2001).
- Lillioja S, Mott DM, Howard BV et al. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N. Engl. J. Med. 318(19), 1217-1225(1988).
- DeFronzo RA. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia. 35(4), 389-397 (1992).
- Shulman GI, Rothman DL, Jue T et al. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 322(4), 223-228 (1990).
- Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann. Intern. Med. 113(12), 909-915 (1990).
- Rothman DL, Magnusson I, Cline G et al. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA. 92(4), 983-987 (1995).
- Cline GW, Petersen KF, Krssak M et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 341(4), 240-246 (1999).
- Goyal R, Faizy AF, Siddiqui SS, Singhai M. Evaluation of tnf-î± and il-6 levels in obese and non-obese diabetics: pre- and postinsulin effects. N. Am. J. Med. Sci. 4(4), 180-184 (2012).
- Saini V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World. J. Diabetes. 1(3), 68-75 (2010).
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 55(s2), S9-9S15 (2006).
- Martin S, Millar CA, Lyttle CT et al. Effects of insulin on intracellular GLUT4 vesicles in adipocytes: evidence for a secretory mode of regulation. J. Cell. Sci. 113(Pt19), 3427-3438 (2000).
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276(42), 38349-38352 (2001).
- Cho H, Mu J, Kim JK et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 292(5522), 1728-1731 (2001).
- George S, Rochford JJ, Wolfrum C et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 304(5675), 1325-1328 (2004).
- White MF. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283(3), E413-422 (2002).
- Whitehead JP, Humphreys P, Krook A et al. Molecular scanning of the insulin receptor substrate 1 gene in subjects with severe insulin resistance: detection and functional analysis of a naturally occurring mutation in a YMXM motif. Diabetes. 47(5), 837–839 (1998).
- Yamauchi T, Tobe K, Tamemoto H et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol. Cell. Biol. 16(6), 3074–3084 (1996).
- Kido Y, Burks DJ, Withers D et al. (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105(2), 199-205.
- Qiao LY, Goldberg JL, Russell JC, Sun XJ. Identification of enhanced serine kinase activity in insulin resistance. J. Biol. Chem. 274(15), 10625-10632 (1999).
- Qiao LY, Zhande R, Jetton TL, Zhou G, Sun XJ. In vivo phosphorylation of insulin receptor substrate 1 at serine 789 by a novel serine kinase in insulin-resistant rodents. J. Biol. Chem. 277(29), 26530-26539 (2002).
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14(18), 1650-1656 (2004).
- Nakae J, Biggs WH 3rd, Kitamura T et al. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 32(2), 245-253 (2002).
- Kitamura T. The role of FOXO1 in β-cell failure and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 9(10), 615-623 (2013).
- Potdar PD, D'souza SB. Isolation of Oct4+, Nanog+ and SOX2- mesenchymal cells from peripheral blood of a diabetes mellitus patient. Hum. Cell. 24(1), 51-55 (2011).
- Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 55(6), 498-510 (2007).
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 51(7), 2005-11 (2002).
- Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Invest. 32 (S3), 14-23 (2002).
- Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney. Int. Suppl. 106, S49-53 (2007).
- Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106(8), 1319-1331 (2010).
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 414(6865), 813-820 (2001).
- Imamura F, O'Connor L, Ye Z et al. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributable fraction. BMJ. 351(h3576) (2015).
- Ozcan U, Cao Q, Yilmaz E et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 306(5695), 457-461 (2004).
- Boffetta P, McLerran D, Chen Y et al. Body mass index and diabetes mellitus in Asia. A cross sectional pooled analysis of 900,000 individuals in the Asia cohort consortium 2011. PLoS. One. 6, e19930 (2011).
- Yoon KH, Lee JH, Kim JW et al. Epidemic obesity and type 2 diabetes in Asia. Lancet. 368(9548), 1681-1688 (2006).
- Willi C, Bodenmann P, Ghali WA, Faris PD, Cornuz J. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 298(22), 2654-2664 (2007).
- Pories WJ, Swanson MS, MacDonald KG et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann. Surg. 222(3), 339-350 (1995).
- Li Y, Ley SH, Tobias DK et al. Birth weight and later life adherence to unhealthy lifestyles in predicting type 2 diabetes: prospective cohort study. BMJ. 351(h3672), (2015).
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes . Dev. 18(18), 2195-2224 (2004).
- Chen F. Is NF-kappaB a culprit in type 2 diabetes? Biochem. Biophys. Res. Commun. 332(1), 1-3 (2005).
- Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 52(5), 1256-1264 (2003).
- Boden G, She P, Mozzoli M et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes. 54(12), 3458–3465 (2005).
- Cai D, Yuan M, Frantz DF et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 11(2), 183-190 (2005).
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, et al. (2005) IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11: 191-198.
- Zou C, Shao J (2008) Role of adipocytokines in obesity-associated insulin resistance. J Nutr Biochem 19: 277-286.
- Mirza S, Hossain M, Mathews C, Martinez P, Pino P, et al. (2012) Type 2-diabetes is associated with elevated levels of TNF-alpha, IL-6 and adiponectin and low levels of leptin in a population of Mexican Americans: a cross-sectional study. Cytokine 57: 136-142.
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102-110.
- Donath MY, Halban PA (2004) Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47: 581-589.
- Moller DE (2000) Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11: 212-217.
- Swaroop JJ, Rajarajeswari D, Naidu JN (2012) Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J Med Res 135: 127-130.
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286: 327-334.
- Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, et al. (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517-1526.
- Liu Y, Biarnés Costa M, Gerhardinger C (2012) IL-1ß Is Upregulated in the Diabetic Retina and Retinal Vessels: Cell-Specific Effect of High Glucose and IL-1ß Autostimulation. PLoS One 7: e36949
- Kuhad A, Chopra K (2009) Attenuation of diabetic nephropathy by tocotrienol: involvement of NFkB signaling pathway. Life Sci 84: 296-301.
- Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, et al. (2004) NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant 19: 2505-2512.
- Lorenzo O, Picatoste B, Ares-Carrasco S, Ramírez E, Egido J, et al. (2011) Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediators Inflamm 2011: 652097.
- Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M (2002) Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 51: 2241-2248.
- Fève B, Bastard JP (2009) The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 5: 305-311.
- Green CJ, Pedersen M, Pedersen BK, Scheele C (2011) Elevated NF-?B Activation is conserved in Human Myocytes Cultured from Obese Type 2 Diabetic Patients and Attenuated by AMP-Activated Protein Kinase. Diabetes 60: 2810-2819.
- Lamson DW, Plaza SM (2002) Mitochondrial factors in the pathogenesis of diabetes: a hypothesis for treatment. Altern Med Rev 7: 94-111.
- DiMauro S, Schon EA (2001) Mitochondrial DNA mutations in human disease. Am J Med Genet 106: 18-26.
- Ingman M, Gyllensten U (2006) mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res 34: D749-751.
- Poulton J, Brown MS, Cooper A, Marchington DR, Phillips DI (1998) A common mitochondrial DNA variant is associated with insulin resistance in adult life. Diabetologia 41: 54-58.
- Samuels DC, Carothers AD, Horton R, Chinnery PF (2006) The power to detect disease associations with mitochondrial DNA haplogroups. Am J Hum Genet 78: 713-720.
- van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, et al. (1992) Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1: 368-371.
- Reardon W, Ross RJ, Sweeney MG, Luxon LM, Pembrey ME, et al. (1992) Diabetes mellitus associated with a pathogenic point mutation in mitochondrial DNA. Lancet 340: 1376-1379.
- Murphy R, Turnbull DM, Walker M, Hattersley AT (2008) Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med 25: 383-99.
- Suzuki S, Oka Y, Kadowaki T, Kanatsuka A, Kuzuya T, et al. (2003) Research Committee or Specific Types of Diabetes Mellitus with Gene Mutations of the Japan Diabetes Society. Clinical features of diabetes mellitus with the mitochondrial DNA 3243 (A-G) mutation in Japanese: maternal inheritance and mitochondria-related complications. Diab Res Clin Pract 59: 207-17
- Tawata M, Hayashi JI, Isobe K, Ohkubo E, Ohtaka M, et al. (2000) A new mitochondrial DNA mutation at 14577 T/C is probably a major pathogenic mutation for maternally inherited type 2 diabetes. Diabetes 49: 1269-1272.
- Wallace DC, Brown MD, Lott MT (1999) Mitochondrial DNA variation in human evolution and disease. Gene 238: 211-230.
- Crispim D, Estivalet AA, Roisenberg I, Gross JL, Canani LH (2008) Prevalence of 15 mitochondrial DNA mutations among type 2 diabetic patients with or without clinical characteristics of maternally inherited diabetes and deafness. Arq Bras Endocrinol Metabol 52: 1228-1235.
- Fainia K (2014) Mitochondrial diabetes. Diapedia 2014
- Liou CW, Chen JB, Tiao MM, Weng SW, Huang TL, et al. (2012) Mitochondrial DNA coding and control region variants as genetic risk factors for type 2 diabetes. Diabetes 61: 2642-2651.
- Ye Z, Gillson C, Sims M, Khaw KT, Plotka M, et al. (2013) The association of the mitochondrial DNA OriB variant (16184-16193 polycytosine tract) with type 2 diabetes in Europid populations. Diabetologia 56: 1907-1913.
- Liu SM, Zhou X, Zheng F, Li X, Liu F, et al. (2007) Novel mutations found in mitochondrial diabetes in Chinese Han population. Diabetes Res Clin Pract 76: 425-435.
- Tang DL, Zhou X, Li X, Zhao L, Liu F (2006) Variation of mitochondrial gene and the association with type 2 diabetes mellitus in a Chinese population. Diabetes Res Clin Pract 73: 77-82.
- Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944-2950.
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, et al. (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54: 8-14.
- Wollheim CB (2000) Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia 43: 265-277.
- Parish R, Petersen KF (2005) Mitochondrial dysfunction and type 2 diabetes. Curr Diab Rep 5: 177-183.
- Widlansky ME, Gokce N, Keaney JF Jr, Vita JA (2003) The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149-1160.
- Sena CM, Pereira AM, Seiça R (2013) Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta 1832: 2216-2231.
- Le Brocq M, Leslie SJ, Milliken P, Megson IL (2008) Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal 10: 1631-74.
- Tabit CE, Chung WB, Hamburg NM, Vita JA (2010) Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev Endocr Metab Disord 11: 61-74.
- Vita JA, Keaney JF Jr (2002) Endothelial function: a barometer for cardiovascular risk? Circulation 106: 640-642.
- Beckman JA, Libby P, Creager MA (2005) Diabetes mellitus, the metabolic syndrome, and atherosclerotic vascular disease. In: Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. (7th edn), WB Saunders, Philadelphia.
- Kim JA, Montagnani M, Koh KK, Quon MJ (2006) Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888–1904.
- Bakker W, Eringa EC, Sipkema P, van Hinsbergh VW (2009) Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res 335: 165-189.
- Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105: 1135-1143.
- Li H, Cybulsky MI, Gimbrone MA, Libby P (1993) An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb 13: 197–204.
- Thorand B, Baumert J, Chambless L, Meisinger C, Kolb H, et al. (2006) Elevated markers of endothelial dysfunction predict type 2 diabetes mellitus in middle-aged men and women from the general population. Arterioscler Thromb Vasc Biol 26: 398-405
- Meigs JB, Hu FB, Rifai N, Manson JE (2004) Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 291: 1978-1986.
- Lee HB, Ha H (2005) Plasminogen activator inhibitor-1 and diabetic nephropathy. Nephrology (Carlton) 10 Suppl: S11-13.
- Ruberg FL, Leopold JA, Loscalzo J (2002) Atherothrombosis: plaque instability and thrombogenesis. Prog Cardiovasc Dis 44: 381-394.
- Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL (2001) Platelet dysfunction in type 2 diabetes. Diabetes Care 24: 1476-1485.
- Navarro-González JF1, Mora-Fernández C (2008) The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433-442.
- Pickup JC (2004) Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 27: 813-823.
- Hu FB, Meigs JB, Li TY, Rifai N, Manson JE (2004) Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes 53: 693-700.
- Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, et al. (2003) Elevated levels of interleukin-18 and tumor necrosis factor-a in serum of patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Metabolism 52: 605-8
- Yang WS, Lee WJ, Funahashi T, Tanaka S, Matsuzawa Y, et al. (2001) Weight Reduction Increases Plasma Levels of an Adipose-Derived Anti-Inflammatory Protein, Adiponectin. J Clin Endocrinol Metab 86: 3815-9.
- van Exel E, Gussekloo J, de Craen AJ, Frölich M, Bootsma-Van Der Wiel A, et al. (2002) Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes : the Leiden 85-Plus Study. Diabetes 51: 1088-1092.
- Liu JJ, Wong MD, Toy WC, Tan CS, Liu S, et al. (2013) Lower circulating irisin is associated with type 2 diabetes mellitus. J Diabetes Complications 27: 365-369.
- Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, et al. (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481: 463-468.
- Catalán V, Gómez-Ambrosi J, Rodríguez A, Silva C, Rotellar F, et al. (2008) Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol (Oxf) 68: 213-219.
- Lancha A, López-Garrido S, Rodríguez A, Catalán V, Ramírez B, et al. (2015) Expression of syntaxin 8 in visceral adipose tissue is increased in obese patients with type 2 diabetes and related to markers of insulin resistance and inflammation. Arch Med Res 46: 47-53.
- Chen MP, Chung FM, Chang DM, Tsai JC, Huang HF, et al (2006) Elevated Plasma Level of Visfatin/Pre-B Cell Colony-Enhancing Factor in Patients with Type 2 Diabetes Mellitus. J Clin Endocrinol Metab 91: 295-299
- Li L, Yang G, Li Q, Tang Y, Yang M, et al. (2006) Changes and relations of circulating visfatin, apelin, and resistin levels in normal, impaired glucose tolerance, and type 2 diabetic subjects. Exp Clin Endocrinol Diabetes 114: 544-548.
- Sreenan SK, Zhou YP, Otani K, Hansen PA, Currie KP, et al. (2001) Calpains play a role in insulin secretion and action. Diabetes 50: 2013-2020.
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, et al. (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26: 163-75.
- Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, et al. (1999) Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 23: 323-328.
- Brunetti A, Chiefari E, Foti D (2014) Recent advances in the molecular genetics of type 2 diabetes mellitus. World J Diabetes 5: 128-140.
- Hoogwerf BJ (2005) Review: complications of Diabetes Mellitus. Int J Dev Countries 25: 63-69.
- Fong DS, Aiello LP, Ferris FL 3rd, Klein R (2004) Diabetic retinopathy. Diabetes Care 27: 2540-2553.
- Aiello LP, Gardner TW, King GL, Blankenship G, Cavallerano JD, et al. (1998) Diabetic retinopathy. Diabetes Care 21: 143-156.
- Nicholson BP, Schachat AP (2010) A review of clinical trials of anti-VEGF agents for diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 248: 915-930.
- Mogensen CE, Christensen CK, Vittinghus E (1983) The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 32 Suppl 2: 64-78.
- Schena FP, Gesualdo L (2005) Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol 16 Suppl 1: S30-33.
- Lee HB, Yu MR, Yang Y, Jiang Z, Ha H (2003) Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 14: S241-245.
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, et al. (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861-9
- Fried LF, Emanuele N, Zhang JH, Brophy M, Conner TA, et al. (2013) Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 369: 1892-903.
- American Diabetes Association (2007) Standards of medical care in diabetes--2007. Diabetes Care 30 Suppl 1: S4-4S41.
- Abbott CA, Malik RA, van Ross ER, Kulkarni J, Boulton AJ (2011) Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 34: 2220-2224.
- Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, et al. (2005) American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 28: 956–962.
- Fowler MJ (2008) Microvascular and Macrovascular Complications of Diabetes. Clinical Diabetes 26: 77-82.
- Backonja M, Beydoun A, Edwards KR, Schwartz SL, Fonseca V, et al. (1998) Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial. JAMA 280: 1831-1836.
- Vinik A (2005) Clinical Review: Use of antiepileptic drugs in the treatment of chronic painful diabetic neuropathy. J Clin Endocrinol Metab 90: 4936-4945.
- Backonja MM (2002) Use of anticonvulsants for treatment of neuropathic pain. Neurology 59: S14-17.
- Max MB, Lynch SA, Muir J, Shoaf SE, Smoller B, et al. (1992) Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N Engl J Med 326: 1250-1256.
- Turner RC, Cull CA, Frighi V, Holman RR (1999) Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 281: 2005-12.
- Kim JJ, Kido Y, Scherer PE, White MF, Accili D (2007) Analysis of compensatory beta-cell response in mice with combined mutations of Insr and Irs2. Am J Physiol Endocrinol Metab 292: E1694-1701.
- Alberti KG, Zimmet PZ (1999) WHO, Geneva, World Health Organization: Definition, Diagnosis, and Classification of Diabetes Mellitus and Its Complications: Report of a WHO Consultation. Part 1: Diagnosis and Classification of Diabetes Mellitus.
- Parsons XH (2014) Direct conversion of pluripotent human embryonic stem cells under defined culture conditions into human neuronal or cardiomyocyte cell therapy derivatives. Methods Mol Biol 1307: 299-318.
- Barclay RS (2015) Combination of Stem Cell and Drug Therapy Could Reverse Type 2 Diabetes.
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, et al. (2008) Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 26: 443–452.
- D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, et al. (2006) Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 24: 1392-1401.
- Cai Q, Bonfanti P, Sambathkumar R, Vanuytsel K, Vanhove J, et al. (2014) Prospectively Isolated NGN3-Expressing Progenitors From Human Embryonic Stem Cells Give Rise to Pancreatic Endocrine Cells. Stem Cells Transl Med 3: 489-499.
- Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, et al. (2001) Insulin production by human embryonic stem cells. Diabetes 50: 1691-1697.
- Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, et al. (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418: 41-49.
- Estrada EJ, Valacchi F, Nicora E, Brieva S, Esteve C, et al. (2008) Combined treatment of intrapancreatic autologous bone marrow stem cells and hyperbaric oxygen in type 2 diabetes mellitus. Cell Transplant 17: 1295-1304.
- Wang L, Zhao S, Mao H, Zhou L, Wang ZJ, et al. (2011) Autologous bone marrow stem cell transplantation for the treatment of type 2 diabetes mellitus. Chin Med J (Engl) 124: 3622-3628.
- Karnieli O, Izhar-Prato Y, Bulvik S, Efrat S (2007) Generation of insulin-producing cells from human bone marrow mesenchymal stem cells by genetic manipulation. Stem Cells 25: 2837-2844.
- Lu D, Chen B, Liang Z, Deng W, Jiang Y, et al (2011) Comparison of bone marrow mesenchymal stem cells with bone marrow-derived mononuclear cells for treatment of diabetic critical limb ischemia and foot ulcer: a double-blind, randomized, controlled trial. Diabetes Res Clin Pract 92: 26–36.
- Si Y, Zhao Y, Hao H, Liu J, Guo Y, et al. (2012) Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes 61: 1616–1625.
- Madec AM, Mallone R, Afonso G, Abou Mrad E, Mesnier A, et al. (2009) Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia 52: 1391-1399.
- Zhao K, Hao H, Liu J, Tong C, Cheng Y, et al. (2015) Bone marrow-derived mesenchymal stem cells ameliorate chronic high glucose-induced ß-cell injury through modulation of autophagy. Cell Death Dis 17: e1885.
- Ho JH, Tseng TC, Ma WH, Ong WK, Chen YF, et al. (2012) Multiple intravenous transplantations of mesenchymal stem cells effectively restore long-term blood glucose homeostasis by hepatic engraftment and ß-cell differentiation in streptozocin-induced diabetic mice. Cell Transplant 21: 997-1009.
- Hao H, Liu J, Shen J, Zhao Y, Liu H, et al. (2013) Multiple intravenous infusions of bone marrow mesenchymal stem cells reverse hyperglycemia in experimental type 2 diabetes rats. Biochem Biophys Res Commun 436: 418-23.
- Heffernan C, Sumer H, Verma PJ (2011) Generation of clinically relevant "induced pluripotent stem" (iPS) cells. J Stem Cells 6: 109-127.
- Soejitno A, Prayudi PK (2011) The prospect of induced pluripotent stem cells for diabetes mellitus treatment. Ther Adv Endocrinol Metab 2: 197-210.
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861-872.
- Jeon K, Lim H, Kim JH, Thuan NV, Park SH, et al. (2012) Differentiation and transplantation of functional pancreatic beta cells generated from induced pluripotent stem cells derived from a type 1 diabetes mouse model. Stem Cells Development 21: 2642– 2655
- Alipio Z, Liao W, Roemer EJ, Waner M, Fink LM, et al. (2010) Reversal of hyperglycemia in diabetic mouse models using induced-pluripotent stem (iPS)-derived pancreatic beta-like cells. Proc Natl Acad Sci USA 107: 13426-13431.