Research Article - Clinical Practice (2017)
Different effects of simvastatin on human airway smooth muscle cell proliferation induced by platelet activating factor
- *Corresponding Author:
- Yi Shen
Changzhou First People’s Hospital,
Changzhou, Jiangsu, China
E-mail: shenyi2010sy@aliyun.com
Abstract
Determine whether and how simvastatin (Sim) may influence human airway smooth muscle cells (HASMCs) proliferation induced by platelet activating factor (PAF) through NADPH oxidase-ROS signaling pathway. Cell proliferation was analyzed according to the protocol of CCK-8 assay. Intracellular reactive oxygen species was measured by the fluorescence using an oxidation sensitive fluorescent probe, 2′7′-dichlorofluorescin diacetate (DCFHDA). The mRNA of Nox4, MnSOD, and Catalase were determined by real-time PCR. Protein levels of Nox4, phospho-ERK, total ERK were determined by western blotting. Inhibitor of MEK1/2was used to confirm the involved signal pathway. Our data demonstrated that PAF induced HASMCs proliferation with the maximal effect seen at 10-6 M and 24 h, the proliferation rate was (1.77 ± 0.51)as fold as the control group. Simvastatin (at the concentration of 10-5 M) was (0.67 ± 0.18), inhibited HASMCs proliferation induced by PAF (P<0.01).Compared with the negative control group (76.79 ± 6.05), PAF-treated group (98.89 ± 1.28) led to a significant increase in DCF fluorescence, PAF combined with simvastatin group (66.40 ± 2.87) decreased ROS level (P<0.01).PAF could increase Nox4 mRNA expression, reduced MnSOD, Catalase levels, simvastatin played opposite role. Treating HASMCs with simvastatin abrogated the phosphorylation of ERK induced by PAF. When added N-acetyl-cysteine (NAC) or PD98059 (ERK inhibitor) to PAF group separately, phosphorylation of ERK reduced at different degree. PAF induced the proliferation of HASMCs through NADPH oxidase-ROS signaling pathway, which was reversed by Simvastatin. Pretreatment of Antioxidant (NAC) significantly reduced ERK phosphorylation. The present study demonstrates ROS dependent activation of ERK/MAPK signaling way.
Keywords
airway smooth muscle cells, simvastatin, reactive oxidant stress, airway remodeling
Introduction
Airway smooth muscle remodeling has been recognized as one of the most important factors that patients fail to respond well to conventional therapy. Airway remodeling in asthma has been recognized as structural changes of airways such as smooth muscle hypertrophy and hyperplasia, thickening and fibrosis of sub-epithelial basement membrane, hypertrophy of bronchial glands, goblet cell hyperplasia, and thickening of airway epithelium. It is widely agreed that a link exists between oxidants and their effect on asthma pathogenesis. Oxidative stress can up-regulate Th2-driven inflammation, thus contributing to increase disease severity, bronchial hyperresponsiveness and airway remodeling [1]. Statins have the effects of antioxidation and have been used for myocardial remodeling. PAF is a proinflammatory phospholipid in the pathogenesis of asthma, which can induce the main characteristics of asthma. Extracellular signal regulated kinases(ERK) are one of the families of Mitogen-activated protein kinase (MAPK) which have shown to be important in the differentiation, activation, proliferation, degranulation and migration of various cells, including airway smooth muscles[2]. The aim of present study was to further assess the role of ERK/MAPK in simvastatin inhibited HASMCs proliferation induced by PAF through NADPH oxidase-ROS signaling pathway.
Materials
Reagents and antibodies
Platelet activating factor (PAF) was purchased from TOCRIS. Simvastatin,2′,7′- Dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma (Paisley, UK), and the ERK inhibitor PD 98059 was purchased from Merck (Beeston, UK). NAC was purchased from biosharp (China). Antibodies against human Nox4, was purchased from Abcam (Cambridge, UK), total ERK and phospho- ERK were purchased from cell signaling technology (USA). Cell counting kit-8(CCK-8) was purchased from Beyotime (China).
Cell culture
HASMCs were obtained from Sciencell (USA). HASMCs were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) (Hyclone, USA) and 1% antibioticantimycotic solution (Gibco, USA).Cells at passages 4 to 8 were used for the experiments.
Cell Proliferation assay
To investigate the effects of PAF on HASMCs proliferation: HASMCs were plated at 5 × 103 cells/well in 96-well dishes and cultured in DMEM supplemented with 10% FBS until they reached 70% confluence. After serum starvation for 24 h, HASMCs were divided into an zero group, an control group, an serum group, an PAF group divided into five subgroups(at different final concentrations of10-5∼10-9 M).Then HASMCs were incubated for 24, 48h, respectively. Add 10 μl of the CCK-8 solution to each well of the plate. Incubate the plate for 1 hour in the incubator. Measure the absorbance (A) at 450 and 620 nm using a microplate reader. A=OD450-OD620.
To investigate the effects of simvastain on HASMCs proliferation: HASMCs were divided into an zero group, an control group, an PAF10-6 M group, Simvastatin10-4 + PAF10-6 M group, Simvastatin10-5 + PAF10-6 M group, Simvastatin10-6 + PAF10-6 M group, Simvastatin10-7 + PAF10-6 M group. Then HASMCs were incubated for 24 h. Add 10 μl of the CCK-8 solution to each well of the plate. Incubate the plate for 1 hour in the incubator. Measure the absorbance at 450 and 620 nm using a microplate reader. A=OD450-OD620.
Measurement of intracellular reactive oxygen species formation
Production of ROS was measured using an oxidation sensitive fluorescent probe, 2′7′-dichlorofluorescindiacetate (DCFH-DA) methods, based on the ROS-dependent oxidation of DCFH-DA to DCF.HASMCs were plated at 1.5 × 105 cells/well in 6-well dishes and cultured in DMEM supplemented with 10% FBS until they reached 70%confluence. After serum starvation for 24 h, HASMCs were divided into a control group (cells without DCFH-DA), negative control group (HASMCs+DCFHDA), PAF group, Simvastatin group, PAF+ Simvastatin group, positive control group (H2O2 × 10-4 M group).Cells were incubated for 24 h. Then incubate with 10 μM DCFH-DA for 20 min in serum-free DMEM. At the end of the incubation period, the DCFH-DA solution was removed and the cells were washed with serumfree DMEM for three times. Fluorescence microscope was used to observe fluorescence intensity of each group.
Divide groups as mentioned above. Centrifugate and harvest cells. After centrifugation at 1000 rpm for 5 minutes, the supernatant was discarded. The pellet was suspended with DCFH-DA (at final concentration of 10 μM). Incubate cells for 30 minutes in the incubator. During the incubating period, blend tubes upside down every 5 minutes, make the probe and cells fully contact. Then, the cells were harvested and washed with PBS for 3 times. After that, suspend in 200 μL PBS. ROS generation was measured by the fluorescence intensity of 10,000 cells with a flow cytometer (BD Company, USA) at excitation wavelength of 488 nm and emission wavelength of 525 nm.
RealTime-PCR
HASMCs were divided into a control group, PAF group, Simvastatin group, PAF+ Simvastatin group. Total RNA was extracted by TRIZOL (Invitrogen, USA). Real-time PCR was performed with the ABI StepOnePlus™ PCR (Applied Biosystems, Foster City, CA) using the SYBRGreenmethod. The primers were purchased from Invitrogen and β-actin served as internal control. The primers used were as follows: Nox4: Sense,5’-CACCAGATGTTGGGGCTAGG- 3’; Antisense, 5’- TGATCCTCGGAGGTAAGCCA- 3’. MnSOD:Sense,5’-AGCAACGGGGACACTTACAA- 3,;Antisense,5’- GTAGTAAGCGTGCTCCCACA-3’. Catalase: Sense,5’-CTGACTACGGGAGCCACATC-3’; A n t i s e n s e , 5 ’ - AGATCCGGACTGCACAAAGG- 3’. β-actin: Sense,5’-ACTGGAACGGTGAAGGTGAC- 3’; Antisense,5’-AGAGAAGTGGGGTGGCTTTT- 3’. All samples were examined intriplicate and included no-template controls.
Western blotting
HASMCs were divided into an control group,PAFgroup,PAF+Simgroup,PAF+ PD98059 (20 μM), PAF+NAC (10 mM). After treatment, cells were washed 2 times with PBS and lysed with Lysis buffer (Keygene, China) containing Protease and phosphatase inhibitors on ice. Protein concentration was measured with the BCA Protein assay kit. Twenty-microgram aliquots of protein were separated by polyacrylamide-8% SDS polyacrylamide gel, and transferred to PVDF membranes. Membranes were blocked with blocking solution (TBST containing 5% BSA) for 120 min at room temperature. Membrane was washed with TBST (TBS containing 0.1% Tween-20) and incubated with phospho-ERK (1:1000), total ERK, (1:1000), Nox4 (1:500) antibody overnight, after washing membranes were incubated with HRP-conjugated secondary antibody for 120 min. The immune complexes were visualized by enhanced chemiluminescence using ECL kit (Amersham Biosciences,Sweden).
Statistical analysis
Results have been expressed as mean ± SEM of 3 independent experiments. Multiple comparisons were made by one-way ANOVA. Results were considered significant at p<0.05.
Results
PAF Induced the Proliferation of HASMCs, which was reversed by Simvastatin
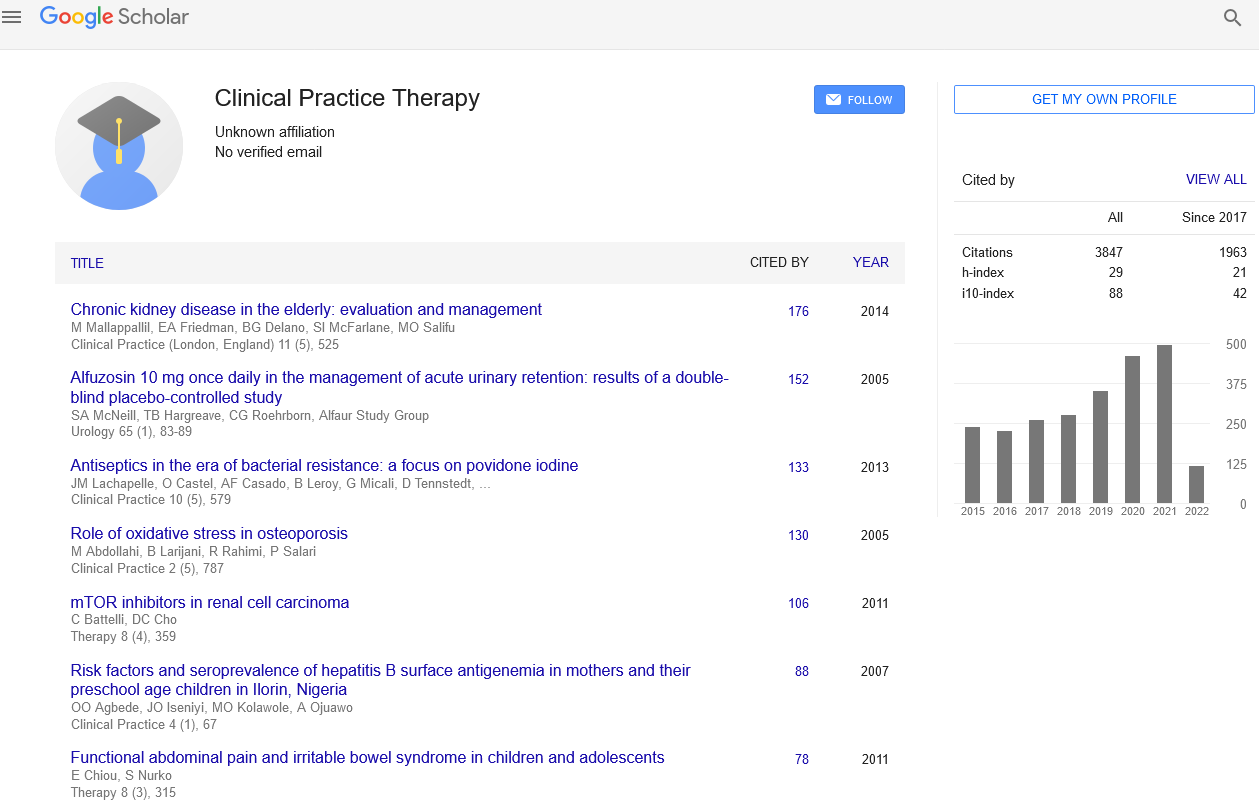
As shown in FIGURE 1, PAF (at different concentration of 10-6 ∼ 10-9 M, 24 hours) significantly increased HASMCs proliferation, with the maximal effect seen at10-6 M, the proliferation rate was (1.77 ± 0.51) as fold as the control group, the difference had statistical significance (p<0.01). PAF10-5M (0.11 ± 0.11) could inhibit HASMCs proliferation, the difference had statistical significance (p<0.01). When treated for 48h (FIGURE 1), compared with the control group, the serum group and PAF group (at different concentration of 10-6 ∼ 10-9 M) significantly increased cell proliferation. Among these, the differences of the serum group (2.36 ± 0.52) and PAF10-6 (1.65 ± 0.25) had statistical significance (p<0.01).The differences of PAF10-7 mol/L (1.42 ± 0.42), PAF10-8 mol/L (1.53 ± 0.13) had statistical significance (p<0.05). PAF10-5 M (0.035 ± 0.019) could inhibit HASMCs proliferation, the difference had statistical significance (p<0.01). PAFtreated group, compared with cells incubation for 48 hours, the cells incubated for 24 hours had more obvious proliferation. We chose 10-6 M as the optimal concentration and 24 hours as the optimal incubation time. Then we investigated the potential effect of simvastatin on cell proliferation (FIGURE 2). Compared with PAF alone (1.16 ± 0.35), Simvastatin (at different concentration of 10-4 ∼ 10-7M) significantly inhibited HASMCs proliferation, with the maximal effect seen at10-5 M, the inhibition rate was 0.67 ± 0.18, difference had statistical significance (p<0.01).

Figure 1. The proliferation rate of HASMCs, compared with control group (24 h), *P<0.01, **P<0.05;compared with control group(48 h), #P<0.01, ##P<0.05.

Figure 2. The inhibition rate of HASMCs, compared with PAF group, * P<0.01, **P<0.05.
PAF increased reactive oxygen species formation, while simvastatin inhibited it
FIGURE 3 showed the images obtained from the fluorescence microscope which showed green fluorescence from cells covered with DCFH-DA indicating the generation of ROS in treatments. (FIGURE 4A and 4B) showed ROS levels measured by mean fluorescence intensity (MFI) with a flow cytometer. Compared with the negative control group (76.79 ± 6.05, PAFtreated groups (98.89 ± 1.28) led to a significant increase in DCF fluorescence, PAF combined with simvastatin group (66.40 ± 2.87) decreased in the intensity of fluorescence. The results showed that simvastatin could decrease the level of ROS in HASMCs induced by PAF. Differences had statistical significance (p<0.01).

Figure 3. Images obtained from the fluorescence microscope showed green fluorescence from cells covered with DCFH-DA.

Figure 4A. ROS levels measured by MFI with a flow cytometer.

Figure 4B. ROS levels of each group,compared with negative control group,*P<0.01, Compared with PAF. group, **P<0.01.
PAF increased Nox4 mRNA expression, reduced MnSOD, Catalase expression, simvastatin played opposite role
Stimulation with PAF for 24 hours (FIGURE 5A-5C) increased Nox4 mRNA expression compared with control group. In contrast with its effect on Nox4 expression, PAF strongly inhibited MnSOD and catalase mRNA. Stimulation with simvastatin alone for 24 hours decreased Nox4 mRNA expression compared with control group and increased MnSOD and catalase mRNA. Stimulation with PAF combined with simvastatin for 24 hours decreased Nox4 mRNA expression compared with PAF alone. While, PAF combined with simvastatin increased MnSOD and catalase mRNA expression, differences all had statistical significance (p<0.01).

Figure 5A. Nox4 mRNA expression, compared with control group,*P<0.01; compared with PAF group, **P<0.01.

Figure 5B. MnSOD mRNA expression, compared with control group,*P<0.01; compared with PAF group, **P<0.01.

Figure 5C. CAT mRNA expression, compared with control group, *P<0.01; compared with PAF group, **P<0.01.
Simvastatin inhibit Nox4, and phosphorylation of ERK expression of HASMCs induced by PAF
Compared with control group (FIGURE 6A and 6B), PAF increased Nox4 protein level. PAF combined with simvastatin or NAC group separately decreased Nox4 level. Compared with control group (FIGURE 7A and 7B), PAF induced ERK1/2 phosphorylation in HASMCs. Treating HASMCs with simvastatin abrogated the phosphorylation of ERK. When added NAC or PD98059 (ERK inhibitor) to PAF group separately, phosphorylation of ERK reduced at different degree. Differences all had statistical significance (p<0.01).

Figure 6A. Nox4 protein expression, compared with control group,*P<0.01; compared with PAF group, **P<0.01.

Figure 6B. Nox4 protein expression.

Figure 7A. ERK phosphorylation expression, compared with control group, *P<0.01; compared with PAF group, **P<0.01.

Figure 7B. ERK phosphorylation expression.
Discussion
Platelet-activating factor (PAF) is one of the most potent and versatile pro-inflammatory mediators. PAF induces vascular permeability, neutrophil and eosinophil chemo taxis, bronchoconstriction, airway hyper reactivity, the metabolism of superoxide and nitric oxide and so on [3].The human PAF receptor (PAFR) is a member of the G protein-coupled receptor (GPCR) family which is expressed on numerous cell types including platelets, monocytes, neutrophils and eosinophils [4]. PAF causes phosphorylation of its receptor, tyrosine phosphorylation of various proteins, activation of phospholipases A2 and C and of phosphatidyl inositol3-kinase and Akt, increased phosphatidylinositol turnover, activation of mitogen activated protein(MAP) kinases and increased cytosolic calcium levels [5].
ROS act as important second messenger molecules, and in vascular tissues may mediate responses such as gene expression, growth, survival/apoptosis, migration, adhesion, inflammation, as well as modulating smooth muscle contractility and endothelial permeability [6-9].The cellular sources of ROS include the peroxisomes and NADPH oxidases [10]. Cellular ROS are maintained at normal levels by enzymatic and nonenzymatic antioxidant mechanisms. MnSOD and catalaseare key enzymatic antioxidants, protecting cells from mitochondrial and peroxisomal ROS, respectively [11]. In our study, we showed that PAF induced the expression of Nox4 while at the same time inhibiting the expression of MnSOD and catalase. These findings suggest PAF induced HASMCs proliferation partly through NADPH oxidase-ROS signaling pathway. Redox regulation’s prime targets include the src family of non-receptor tyrosinekinases (srcFK), protein kinase C, and mitogen-activated proteinkinases (MAPKs), Rho-kinase and, so on [12]. MAPKs consist of three families: ERK, p38 MAPK (p38), and jun N-terminal kinase. All MAPKs are activated through a signaling cascade starting with activation of the small G proteins including: ras, rac-1, or cdc42, ending with dual tyrosine/threonine phosphorylation of the MAPK [13]. ERK has also been implicated in control of vascular function in response to cellular stresses such as hypoxia, oxidative stress, and inflammation, as well as to vasoconstrictor hormones [14].However, whether oxidative stress via ERK1/2 MAPK pathways in HASMCs has not been reported. Our data showed that PAF induced ERK1/2 phosphorylation in HASMCs partly support our proposition. To investigate the effect of a MAPK/ERK kinases (MEK) inhibitor on the induction of ERK1/2 phosphorylation by PAF, cells were pretreated with PD98059.Our data showed pretreatment with PD98059 abolished ERK phosphorylation elicited by PAF, suggesting that ERK phosphorylation in the presence of PAF is MEK-dependent.
In our experiment, simvastatin could inhibit HASMCs proliferation through NADPH oxidase-ROS signaling pathway. The underlying mechanisms are as follows. Statins can inhibit 3-hydroxy3-methylglutaryl coenzyme A (HMG-CoA) reductase the rate limiting enzyme for synthesis of mevalonic acid, a precursor for cholesterol, farnesyl and geranylgeranyl pyrophosphate isoprenoids. Posttranslational farnesyl or geranylgeranylation of low molecular weight GTP-binding proteins such as RAS and RHO are thought to be an essential step in activation of phosphorylation cascades such as the RAS-RAF-1-MEK-1-MAPK/ERK pathway which stimulate cell proliferation [15]. N-acetylcysteine is the acetylated variant of the amino acid L-cysteine. Its supplementation supported by scientific evidence include prevention of chronic obstructive pulmonary disease exacerbation, treatment of pulmonary fibrosis, etc. [16]. In our study, when added NAC to the cultures, the phosphorylation of ERK1/2 were sharply reduced. It demonstrates antioxidant could not only protect the body from direct damage from various kinds of oxidants, but also prevent airway remodeling.
Oxidative stress is the final result of most of the molecular pathways and mechanisms in asthma, which can result in irreversible damage and form airway remodeling. Statins have the effects of inhibiting inflammation and antioxidation. This experiment provides experimental basis for statins in the treatment of airway remodeling. Statins may represent a potentially new approach in the treatment of asthma.
References
- Frossi B, De Carli D, Piemonte M, et al. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cell. Mol. Immunol.45, 58-64 (2008).
- Duan W, Wong WS. Targeting mitogen-activated protein kinases for asthma. J. Curr. Drug Targets 7(6), 691-698 (2006).
- Salluh JI,Pino AV,Silva AR, et al.Lung production of platelet-activating factor acetylhydrolase in oleic acid-induced acute lung injury. Prostaglandins Leukot. Essent Fatty Acids 77(1), 1-8 (2007).
- Dupré DJ, Rola-Pleszczynski M, Stankova J.Rescue of internalization-defective platelet-activating factor receptor function by EBP50/NHERF1. J. Cell Commun. Signal. 6(4), 205-216 (2012).
- Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutantmice. Prog. Lipid Res. 39, 41-82 (2000).
- Ahsan MK, Lekli I, Ray D, Yodoi J, Das DK. Redoxregulation of cell survival by the thioredoxin superfamily:an implication of redox gene therapy in the heart. Antioxid. Redox Signal 11, 2741-2758 (2009).
- Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 20, 2175-2183 (2000).
- Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal 11, 2985-3011 (2009).
- Satoh K, Nigro P, Berk BC. Oxidative stress and vascular smooth muscle cell growth: a mechanistic linkage by cyclophilin A. Antioxid. Redox Signal 12, 675-682 (2010).
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature 408, 239-247 (2000).
- Moldovan L, Moldovan NI. Oxygen free radicals and redox biology of organelles. Histochem. Cell Biol. 122, 395-412 (2004).
- Ward JP, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone-role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol. Ther. 104, 207-231 (2004).
- Qi M , Elion EA. MAP kinase pathways. J. Cell Sci. 118, 3569-3572 (2005).
- Frank GD, Eguchi S, Yamakawa T, et al. Involvement of reactive oxygen species in the activation of tyrosine kinase and extracellular signal regulated kinase by angiotensin II. Endocrinology 141, 3120-3126 (2000).
- Johnson MD, Woodard A, Okediji EJ, et al. Lovastatin is a potent inhibitor of meningioma cell proliferation: evidence for inhibition of a mitogen associated protein kinase. J. Neurooncol. 56(2), 133-142 (2002).
- Millea PJ. N-acetycysteine: multiple clinical applications. J. Am. Fam. Physician. 80(3), 265-269 (2009).